Applications of Metabolic Flux Analysis (MFA)
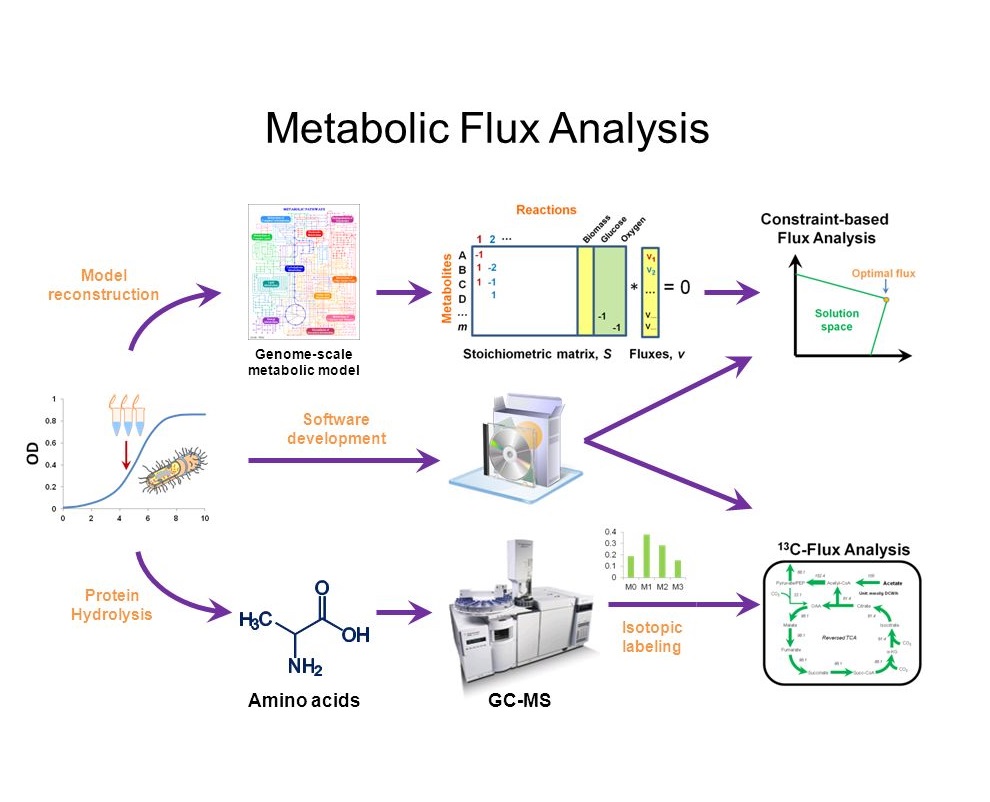
Metabolic Flux Analysis (MFA) is a technique used to quantify metabolic fluxes (the rate of turnover of molecules). MFA has at least two important applications: First, by studying metabolic flux, it is possible to adjust the amount of substrates/ingredients in the medium/protocol of cell culture [1] or mutate genes [2] to improve productivity. Second, by adjusting the diet to maximize the biomass of gut microbiome [4][5], people may achieve better health.
Here are three examples to show the applications of MFA:
- Building the model to predict productivity of monoclonal antibody given different substrate/medium [1].
This study built the model by curating the metabolic network (Figure 1). After the network is curated, the network can predict the output fluxes of monoclonal antibody (mAb) given the input fluxes of nutrients including amino acids, glucose and ammonium. Figure 2. shows the experimental (in symbol) and prediction (in lines) results. Using different medium (Biogro-CHO medium or PowerCHO2 medium) or different culture approach (fed-batch or batch culture) generates different productivity of mAb.
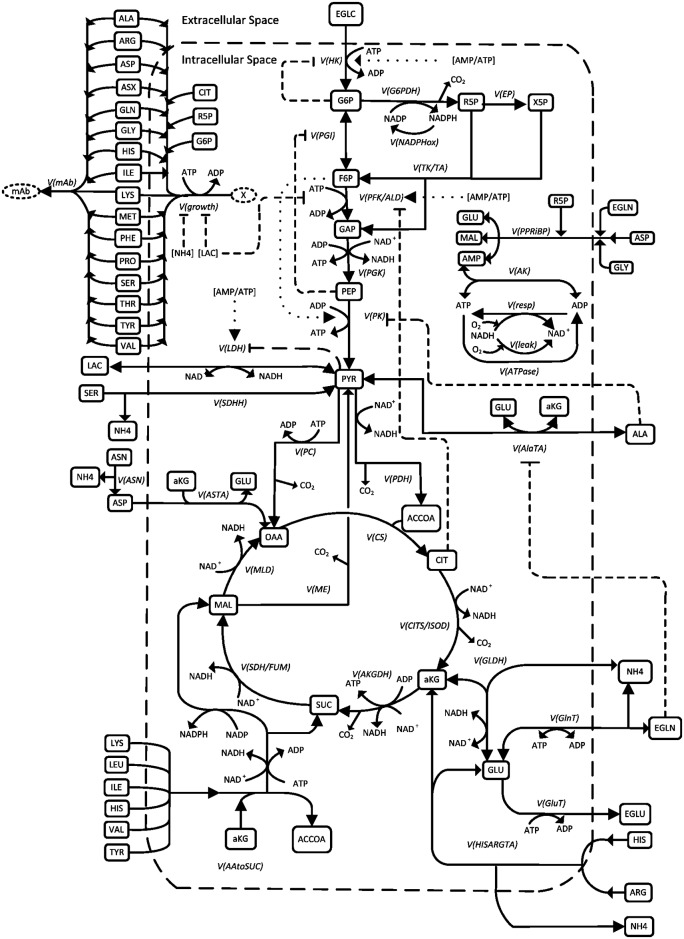
Fig. 1. Metabolic network in Chinese hamster ovary (CHO) cells [1]. By quantifying metabolic fluxes, we can optimize the productivity of product by adjusting the substrates or medium. In this case, product is monoclonal antibody (mAb) and the substrates are amino acids, glucose and ammonium.
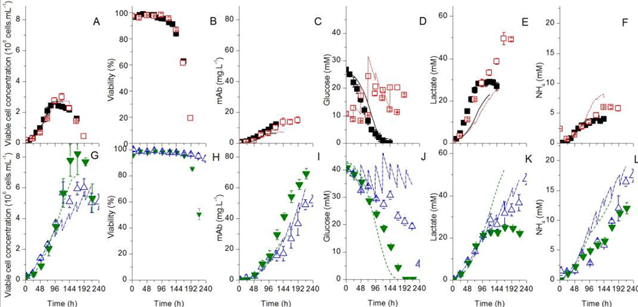
Fig. 2. Time series of variable cell concentration, cell viability, product (mAb) and substrates [1]. The top is the results using Biogro-CHO medium, and the lower part are the results using PowerCHO-2 medium. The filled symbols represent batch culture and the empty symbols represent fed-batch culture.
After building the model that can predict mAb productivity, it is possible to optimize productivity by adjusting the substrates or ingredients of the medium.
- Metabolic fluxes comparisons between wildtype and aroA/aroD genes overexpress mutant.
This study compared the metabolic fluxes comparisons between wildtype and aroA/aroD genes overexpress mutant given the information that this mutant will result in a significant accumulation of product, shikimic acid (SA)[3]. By comparing the fluxes between mutant and wildtype strains, we can know more about the mechanism of SA production, which may be helpful for further SA productivity improvement.
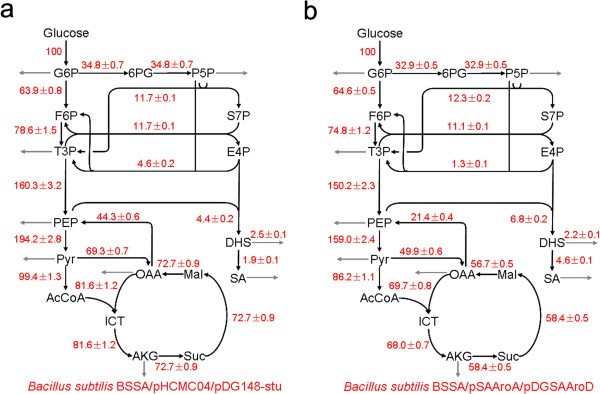
Fig. 3. Shikimic acid (SA) is a commercial product. The strain which overexpresses the aroA and aroD genes (figure 3b.) has a higher SA production rate [2].
- Maximizing the biomass of gut microbiome to improve health status by adjusting diet.
There are studies showing that healthier people have a higher bacterial gene count inside their gut compared with unhealthier people [4]. We assume gene count is correlated with the amount/biomass of gut microbiota – higher gene count may improve the health status. Under these assumptions, an optimization algorithm was proposed to maximize the biomass of microbiome given the flux constraints by adjusting the diet [5]. Finally, eight essential amino acids consumed by the gut microbiome which may increase/decrease the number of microbiomes can be seen in Figure 5 [5].
Fig. 4. Trying to increase biomass of gut microbiome by adjusting the diet: The metabolic pathway between host and microbiome are obtained and then the substrates which can maximize the biomass of microbiome are found. Finally, the food containing these ingredients may be helpful in maximizing the biomass of microbiome.
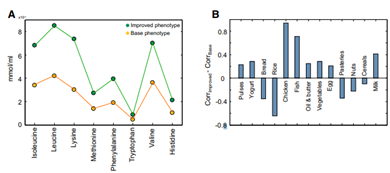
Fig. 5. (A) Simulated consumption of the eight essential amino acids by the gut microbiome of the LGC (yellow circles, unhealthy) individuals and the HGC (green circles, healthy) individuals. (B) Positive/Negative effect of different food sources improve the phenotype of LGC subjects [5].
References: